
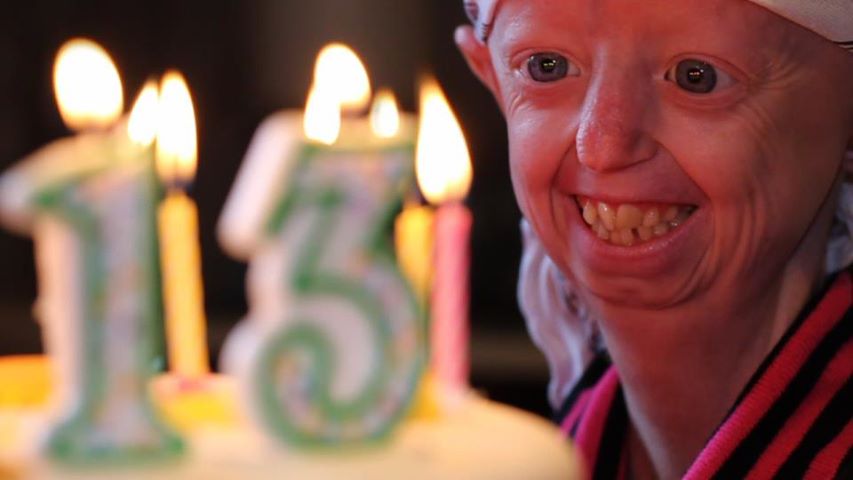
La lucha contra el síndrome de envejecimiento acelerado de Hutchinson-Gilford, un trastorno muy inusual que aparece en la infancia de los dolientes y que causa su muerte prematura, ha podido dar un paso de gigante gracias a un equipo de científicos de la Universidad de Oviedo y del Instituto Salk de California (EE UU).
Según lo informó el portal de 20 minutos, el síndrome de esta enfermedad «ultra-rara» lo produce una mutación en el gen LMNA (uno de los 20.000 que constituyen la formación de una célula) que desencadena la acumulación de una proteína tóxica en el núcleo que provoca finalmente la progeria.
Actualmente, existen tratamientos basados en inhibridores de la farnesiltransferasa y en aminobifosfonatos, que «disminuyen la toxicidad de la proteína que causa esta enfermedad, pero no evitan que se produzca», explica a 20minutos la investigadora española Olaya Santiago, -Fernández, primera autora del estudio.
«Con esta terapia podemos conseguir efectos permanentes porque únicamente es necesario administrar este tratamiento una vez».
Aumenta un 25% la esperanza de vida Liderados por los profesores Carlos López-Otín y Juan Carlos Izpisúa-Belmonte, los científicos han diseñado dos terapias similares basadas en la técnica de edición genética con el sistema CRISPR/Cas9.
«Este sistema ya había sido ensayado previamente para otras enfermedades genéticas, pero esta es la primera vez que se utiliza en una enfermedad sistemática, que afecta a múltiples tejidos», apunta Santiago-Fernández, que ha colaborado en su diseño para que reconociera el gen que se encuentra mutado en los pacientes con envejecimiento acelerado. Un total de 119 pacientes diagnosticados, enumera.
Este estudio, que se ha publicado en dos artículos independientes en la revista Nature Medicine y en el que han trabajado 9 personas durante un periodo de tres años, ha ensayado el sistema en un modelo murino de la enfermedad generado previamente para estudiar la patología, lo que ha permitido prolongar un 25% la vida de los ratones afectados.
Uno de los investigadores del equipo es Sammy Basso, probablemente el paciente con progeria más longevo del mundo con 22 años, que, al igual que Olaya Santiago-Fernández, se interesó en las investigaciones del catedrático Carlos López-Otín. Tras graduarse en Biología Molecular para poder investigar sobre su propia enfermedad, Basso se trasladó al laboratorio para unirse al proyecto y ayudar en sus experimentos.
Las investigaciones en el bioterio de la Universidad y los experimentos in vitro realizados con cultivos celulares en el laboratorio nos han proporcionado «una herramienta capaz de atacar la causa última de la enfermedad, actuando directamente sobre la mutación que la produce», apunta Santiago-Fernández.
«Hemos demostrado que es una posibilidad real, incluso para una enfermedad tan compleja que afecta a múltiples órganos y tejidos del organismo», afirma la investigadora de la institución asturiana.
«Pero debemos ser muy cautelosos para no hacer concebir expectativas poco fundadas a las personas que sufren este tipo de trastornos, puesto que serán necesarios todavía muchos estudios adicionales para poder trasladar con garantías un tratamiento».
El equipo de científicos ha obtenido unos resultados con el modelo de ratones «muy alentadores», produciendo «continuos avances en el sistema CRISPR/Cas9 que aumentarían su eficacia y su especificidad».
La investigadora asegura que son «optimistas acerca de la posibilidad de que se logren avances espectaculares», pero todavía es necesaria «una buena dosis de esfuerzo e investigación en múltiples campos» para lograr un tratamiento definitivo.